关注罕见病⑧|Fabry病
“Fabry!他果然是Fabry病!”主治医师拿到患者符某的肾活检病理、基因及酶检测结果后,为最终能够给患者明确诊断感到欣慰,同时也为科室专业团队的精湛诊疗技术而感到自豪。
几个月前,此患者在当地医院就诊于多个科室,先后被诊断为“充血性心力衰竭”、“胆汁反流性胃炎”、“肾功能不全”,困惑不已的患者辗转至柳州市工人医院肾内科住院。入院经详细病史询问,患者不经意的一句“怕热、少汗”引起经验丰富的肾科大夫警惕,难道是Fabry病?经同患者沟通后,及时为其安排了肾活检。
等待病理结果的同时,谭鹤长主任、黄雯静副主任医师对患者家系进行详细调查:患者的女儿、患者姐姐的女儿、患者妹妹的儿子都有不同程度“皮肤瘙痒、皮炎、怕热、手足神经痛”等症状。男性患者传女不传男,女性患者男女都传,风险各一半,似乎离真相越来越近,后肾活检病理回报:光镜见肾小球足细胞空泡变性,电镜见部分足细胞次级溶酶体增多并大量髓样小体和斑马小体,需结合临床排除Fabry肾病。
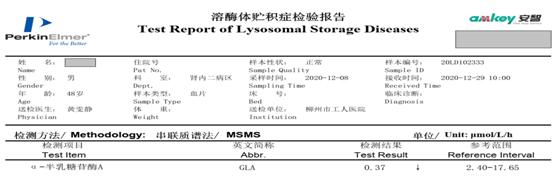
经问诊、体格检查及完善我院能开展的检查,高度提示Fabry病,然而酶及基因检测是金指标,此患者为贫困人口,无法承受昂贵的检测费用,孟晓燕副主任第一时间联系了上海交大瑞金医院肾内科陈楠教授团队,瑞金医院肾内科构建有全球最大的单中心Fabry病家系库,成功为患者争取到免费基因及酶学检测的机会,结果提示酶活性下降、底物升高及可疑致病半合子变异,最终诊断我院首例Fabry病。

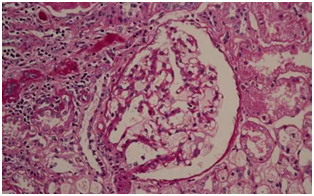
光镜PAS染色见足细胞空泡变性
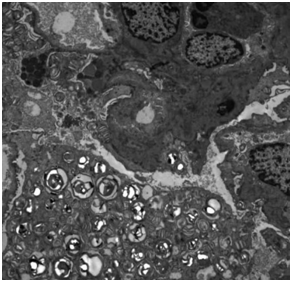
电镜见足细胞大量斑马小体



什么是法布里病?

Fabry病(Fabry disease)的中文译称法布里病,是一种罕见的X连锁遗传性疾病,由于X染色体长臂中段编码α-半乳糖苷酶A(α-Gal A)的基因突变,引起α-Gal A结构和功能异常,导致使其代谢底物三已糖神经酰胺(GL-3)和相关鞘糖脂在全身多器官内大量堆积所导致的临床综合征。属于溶酶体蓄积病,可导致肾功能衰竭、心肌肥厚、四肢神经痛、角膜涡轮状浑浊等严重临床综合征,最终造成肾脏、心脏、神经等多系统的不可逆损伤。
由于临床表现多样、早期症状缺乏特异性、临床认识不足、就诊科室分散等原因,法布里病的诊断极其困难,延迟诊断可长达20年,如患者有以下临床表现,要警惕Fabry病:
面容
男性患者多在12-14岁出现特征性面容,表现为眶上嵴外凸,额部隆起和嘴唇增厚。
皮肤血管角质瘤
常见于经典型患者,多见于“坐浴区”即脐膝之间的外生殖器、阴囊、臀部和大腿内侧,凸出皮肤表面的红色斑点。
神经系统
多数患者会出现周围神经疼痛,表现为足底和手掌难以忍受的烧灼感,并放射到四肢近端,甚至出现痛性痉挛;自主神经受累时表现为少汗或无汗;中枢神经系统受累时多表现为早发的短暂性脑缺血发作或缺血性卒中。
眼
特征性的表现包括结膜血管迂曲、角膜涡状混浊、晶状体后囊混浊和视网膜血管迂曲,严重者可导致视力降低甚至丧失。常为女性患者就诊的主要原因之一。
消化道
多在进食后出现恶心、呕吐、腹胀、痉挛性腹痛和腹泻等症状,也可表现为吸收不良或便秘。
肾脏
早期表现为尿浓缩功能障碍如夜尿增多、多尿和遗尿,随病程进展可逐渐出现蛋白尿,甚至达肾病综合征水平,伴随肾功能损害。
心血管系统
可表现为高血压、冠状动脉受累导致的心肌缺血、心脏瓣膜病变和肥厚性心肌病,严重者可表现为心绞痛、心肌梗死和心力衰竭。多为疾病的晚期表现和主要的死亡原因。
在众多罕见病中,难诊、难治的Fabry病有明确的酶替代治疗方法,酶替代治疗可以在不可逆器官损害前提供有效的干预机会。近年酶替代治疗药物在我国正式引进,2020年5月18日上海瑞金医院肾内科开具了我国首个Fabry病酶替代治疗处方,使得Fabry病成为国内为数不多的可防可治的罕见病之一。
罕见病,是指那些发病率极低的疾病,为患病人数占总人口的0.065%至0.1%的疾病。目前已经明确的罕见病有7000 多种,其中80%为遗传病,如白化病、血友病等,95%的罕见病仍没有特效药。截止2018年5月,我国罕见病患者已达1680多万。作为全国罕见病诊疗协作网成员医院之一,我院在柳州市工人医院智慧医院特设疑难罕见病线上门诊。本门诊有两名主治医师长期在线,主要负责疑难病例资料的收集及初步筛查等工作。有诊断不清的疑难病历均可在该门诊咨询及求助。疑难罕见病线上门诊收集病例后会负责组织全院力量帮助明确诊断,必要时可请上级医院专家线上会诊帮助诊疗。
扫一扫 手机端浏览
 医患交流
医患交流
 微信公众号
微信公众号
 智慧医院
智慧医院